Flexible
Docking with MORDOR
(MOlecular Recognition with
a Driven dynamics OptimizeR)
Major Limitation of Most Existing
Methods.
Treating the receptor as fixed during docking is
generally a necessary evil. In many docking programs, the receptor is
fixed for all calculations. The need to account for the dynamic
behavior of a receptor has long been recognized as a complicating
factor in computational drug design. The energy landscape of most
proteins and nucleic acid are frequently described in terms of a
folding funnel in which there are many highly unfavorable states that
collapse via multiple routes entailing favorable folded states
generally arriving at a single native structure, which still may
possess some conformational variability at ambient temperature. A
single structure, even the weighted average provided by a crystal
structure or NMR structure, may not adequately describe these substates
required for ligand recognition. It is important to note that a
conformational funnel and the states that it represents are
condition-dependent. Altering the conditions (ionic strength, pH,
temperature) changes details of the funnel. One of the most important
points to consider is that introducing a ligand into the system also
changes the environment. It may affect the most populated state of the
receptor; such a case would correspond to an “induced-fit” of ligand
and receptor. The macromolecule most likely exists in a full complement
of conformations: most in the native state, some in the induced-fit
state, and some in other states. If the ligand binds preferentially to
the induced-fit state with sufficiently favorable free energy change,
i.e., greater than the free energy difference between the native and
induced-fit states of the macromolecule, the average structure of the
macromolecule will change. If the ligand is capable of multiple binding
modes to a macromolecule (binding to different folded forms or multiple
binding sites of a single form), it may be necessary to account for
these additional states to predict affinity properly.
Many docking
algorithm build the drugs “in situ”
in
a pocket without
checking how the drugs bind there kinetically. Studies in our lab using
MORDOR show that ~20% of the drugs found with these techniques cannot
reach the pocket. While the “fix” of building “in situ” is certainly an
appropriate response to get calculated structures to match
experimental, it illustrates the difficulty of adequately testing a
potential ligand for binding when the receptor changes its structure in
response to the ligand’s presence.
MORDOR docking
software was primarily designed for accuracy rather than
rapidity of docking. (We will subsequently streamline MORDOR, so it can
run as fast as possible to achieve its goal.) Most importantly, MORDOR
allows induced-fit. 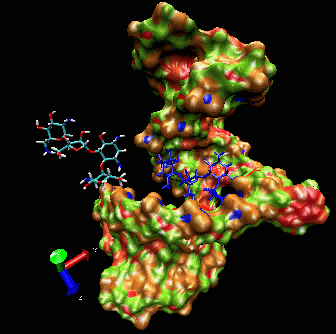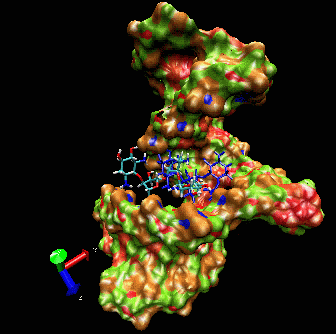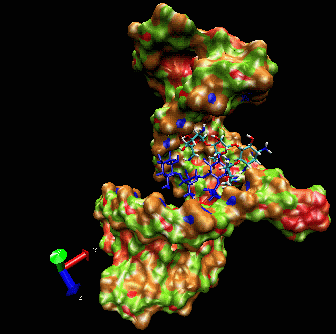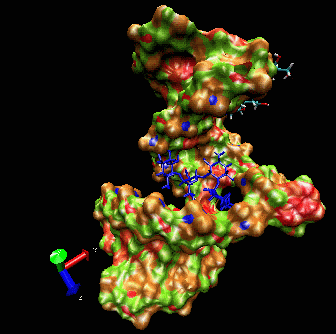 The
ligand is able to displace bases, and it can
create a new bulge or stabilizing/ordering loop of the RNA. For that
purpose, MORDOR implements a novel algorithm for conformational space
search that allows an efficient systematic search during docking. This
approach combines molecular minimization or molecular dynamics
simulation with a driving force
that moves the ligand. Starting from a random position of the ligand
around the receptor, the ligand explores the receptor surface by an
additional root-mean-square-deviation type of force (Path Exploration
With Distance Constraints or PEDC method (see
publications), which constrains the drug to explore the
conformational
space following a low energy pathway. The ligand explores and
identifies all potential binding pockets as shown above.
The
ligand is able to displace bases, and it can
create a new bulge or stabilizing/ordering loop of the RNA. For that
purpose, MORDOR implements a novel algorithm for conformational space
search that allows an efficient systematic search during docking. This
approach combines molecular minimization or molecular dynamics
simulation with a driving force
that moves the ligand. Starting from a random position of the ligand
around the receptor, the ligand explores the receptor surface by an
additional root-mean-square-deviation type of force (Path Exploration
With Distance Constraints or PEDC method (see
publications), which constrains the drug to explore the
conformational
space following a low energy pathway. The ligand explores and
identifies all potential binding pockets as shown above.
One of the most important aspects of this
approach
is that we naturally
take into account "induced-fit" when the drug binds to the receptor.
Movements of side-chains, regions, or even whole domains may be
permitted by the docking procedure. In particular, the presumed binding
pocket may be allowed to change its shape to a certain extent for a
better binding interaction. This allows us to include in our scoring
function the energy change of the macromolecule while changing its
conformation due to the drug interaction. This is very important and
has been neglected until now in drug design. Our preliminary data show
that contributions of the target induced-fit energy change could make
the difference for predicting the correct binding energy. The second
major advantage of using MORDOR is that we have a complete surface map
of where the ligands may bind. That enables assessment of drug
specificity
Flexible
Docking with MORDOR. Studying induced fit with MORDOR is
particularly important in docking protein and particularly nucleic
acids. In fact, nucleic
acid-drug complex structures indicate that drugs do not generally bind
a canonical form of RNA or DNA. On the contrary, known structures of
drugs bound to nucleic acids indicate that very often, the drugs
displace bases thus provoking a local reorganization of the nucleic
acid. Flexible docking for RNA will greatly enhance the probability of
success in a drug discovery program.
Docking Procedure. We have created
and currently use a database of ~400,000 – 500,000 compounds
incorporating the Available Chemicals Directory (ACD) (≥200,000
compounds), the National Cancer Institute directory (~200,000
compounds), and compounds from commercial sources made available to us
(Maybridge, Comgenex, TRC). AMBER/AnteChamber
atom types were assigned based on local connectivity. The partial atom
charge was calculated using the AM1-BCC correction factor.
We first perform
rigid docking using DOCK
effectively to reduce the size of libraries, while retaining the
potentially good drug candidates. DOCK is fast and convenient
to select 10,000 compounds from the database. 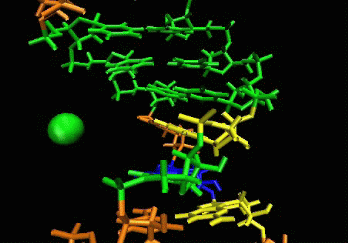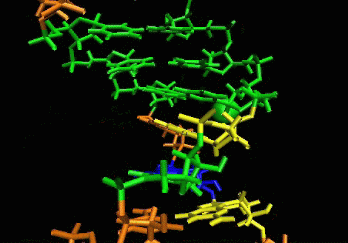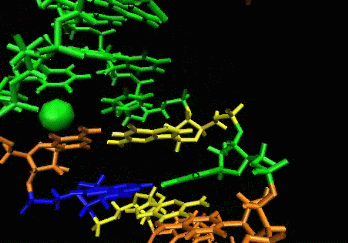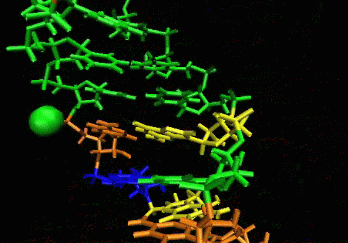 It uses spheres that are supposed to represent the best
pocket on the receptor to fit the ligand. The spheres are usually
generated using accessible surface criteria, where a buried pocket
inside the macromolecule seems to be a place of choice. However, this
rapid approach uses a rigid structure, which precludes discovery of a
pocket made by the receptor as it alters its conformation due to the
presence of the ligand. To find new hot spots, we developed a procedure
using MORDOR to calculate the binding interaction energy map between a
receptor and a dummy sphere (given a VDW radius and a charge). The
dummy sphere is placed and moved along a grid that covers the entire
receptor. Both receptor and dummy sphere are flexible and are minimized
simultaneously. The result is an interaction energy map between the
receptor and the dummy sphere that identifies and finds interesting new
hotspots to interact with ligands. This procedure was able to identify
all known pockets in our training test set among the top ten spheres,
i.e., those that exhibited the best interaction energy with the
receptor. Sphere generation using MORDOR is illustrated above.
It uses spheres that are supposed to represent the best
pocket on the receptor to fit the ligand. The spheres are usually
generated using accessible surface criteria, where a buried pocket
inside the macromolecule seems to be a place of choice. However, this
rapid approach uses a rigid structure, which precludes discovery of a
pocket made by the receptor as it alters its conformation due to the
presence of the ligand. To find new hot spots, we developed a procedure
using MORDOR to calculate the binding interaction energy map between a
receptor and a dummy sphere (given a VDW radius and a charge). The
dummy sphere is placed and moved along a grid that covers the entire
receptor. Both receptor and dummy sphere are flexible and are minimized
simultaneously. The result is an interaction energy map between the
receptor and the dummy sphere that identifies and finds interesting new
hotspots to interact with ligands. This procedure was able to identify
all known pockets in our training test set among the top ten spheres,
i.e., those that exhibited the best interaction energy with the
receptor. Sphere generation using MORDOR is illustrated above.
For present purposes
of development, at the last
stage of docking, the top 1000 compounds will be selected and
investigated further for additional criteria: entropy term in the
scoring function, drug specificity estimation by checking if the drugs
can also bind somewhere else to the RNA surface, pathway of the ligand
to reach the pocket from the solvent to assess whether it is
kinetically favorable.
Features of MORDOR.
- Flexible docking simulations using
minimization or molecular
dynamics optimization in Cartesian space. The ligand explores the
receptor surface using the PEDC (see publications).
- Ligands and receptor are
flexible. This permits
“ induced-fit” when the drug binds to the target, allowing
exploration of other interesting binding sites which were buried in the
original structure (X-ray or NMR).
- Use CHARMM or AMBER force field for the
receptor and Antechamber/AM1-BCC
for the ligand. We have all of these.
- Procedure to generate a complete
surface map landscape where the
drugs bind around the protein or nucleic acid. This has a bonus of
permitting us to assess ligand specificity.
- An unlimited number of constraints
or restraints are allowed,
giving greater control over docking. This allows any part of the ligand
and receptor to be automatically constrained to a pre-defined position
during docking.
- The pathway used by the ligand to
reach the pocket from the
solvent can be examined to gain insight or assess feasibility.
- Identify the best binding receptor
by probing the surface of the
macromolecule with a dummy sphere with user-selected VDW radius and a
charge.
- Written in C++.
- Platforms Available: Linux, windows.
- Scripting in Python.
MORDOR
Development. We are working on several aspects of docking.
MORDOR needs a very fast computational resource. It quickly becomes a
very slow process if screening a million compounds from vendor
databases or in-house libraries. It takes about 1-10 CPU-hr per complex
to generate the PEDC-driven pathway of binding using our current small
cluster. The proposed new cluster makes it possible to screen more than
10,000 compounds with MORDOR in its current non-optimized form.
We are also developing a new, particularly fast procedure using MORDOR
to pre-screen the database (instead of using DOCK), by predicting
binding affinity rapidly and with a reasonable level of accuracy.
We need to make
improvements in the accuracy of
the scoring function to enhance the reliability of discriminating
correctly docked from misdocked conformations by adding:
- A solvation energy term using one of
the methods discussed in the
section on DOCK (Generalized Born, linearized Poisson-Boltzmann, and
non-linear Poisson-Boltzmann)
- An entropy term which will be
derived from a quasinormal mode
calculation. These will entail comparative studies, since we will need
accuracy tempered by the time involved for a calculation.