Installation
Windows
We provide 32 bits and 64 bits installers. Choose the one that corresponds to your operating system, double click on it and follow the installer instructions. The installer will install both the MCR and PhenoRipper.
Mac OSX
We provide a Mac OSX compiled version of PhenoRipper. Due to MATLAB restrictions, the compiled code will only work on Mac OSX versions ≥ 10.6.
1/ Install the MCR (Matlab Compiler Runtime)
- Double clicking on the file MCRInstaller.zip
- It will unzip the MCR into the folder MCRInstaller. Go to this folder
- Double click on the file InstallForMacOSX to sart the MCR installer
- If possible, install it in the default folder ( /Applications/MATALB/MATLAB_Compiler_Runtime/v716)
2/ Install PhenoRipper
- Double click on the file PhenoRipper.zip to unzip the disk image
- Double click on the file PhenoRipper.dmg to mount the disk image
- Drag the folder phenoripper into the Applications folder
- To start PhenoRipper, if you used the default path for the MCR installer, double click on the phenoripper.command file inside the phenoripper folder.


Linux
Installation on GNU/Linux is not available yet, but it will be supported soon (by 6/12).
Start
Windows
Double click on the icon PhenoRipper
 and wait to see the main interface.
and wait to see the main interface.
Mac OSX
If the MCR has been installed using the default setting
- Go, using Finder, to the folder Applications>PhenoRipper
- double click on the file phenoripper.command

If the MCR has NOT been installed using the default setting
You can always edit this file and change it if the path to the MCR is different on your computer.
You can also run PhenoRipper using the Terminal application: start your Terminal application located in Application>Utilities folder. Once launched, Terminal opens a single window displaying a greeting and a second line of text shown in the figure below:

You will have to use the Terminal to go to the PhenoRipper folder. Enter:
cd /Applications/PhenoRipperThen start the PhenoRipper application by using the following command:
./run_gui.sh /Applications/MATLAB/MATLAB_Compiler_Runtime/v715/and wait to see the main interface.
PhenoRipper Main Interface
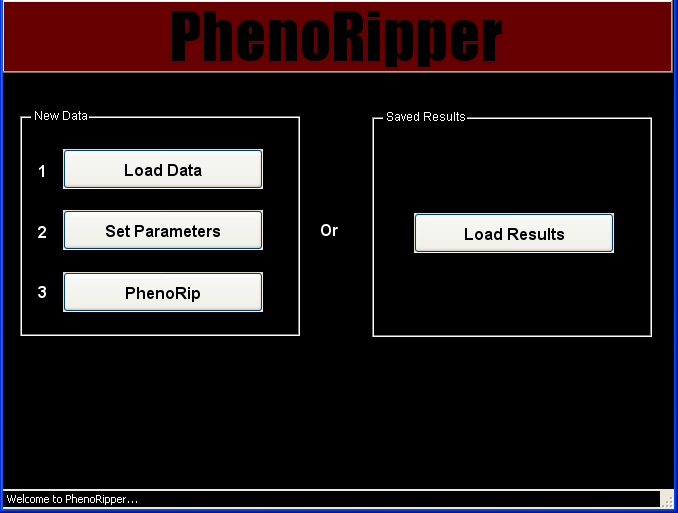 PhenoRipper main interface allows you to:
PhenoRipper main interface allows you to:
- Load data
- Load previously “PhenoRipped” results
Let’s first see how to load new data into PhenoRipper.
Load Data
To load some data, press the Load Data button.
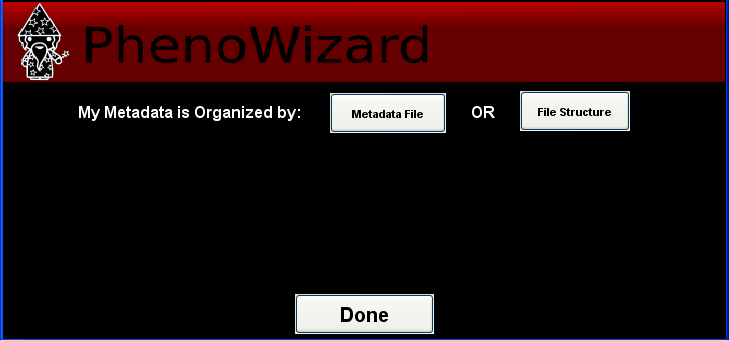
There are two ways to load data using PhenoRipper: using a metadata file or by extracting the file information from the file path. In this tutorial, we will load the data using the Metadata File.
Using Metadata File
Click on Metadata File and select the metadata file provided with the example.

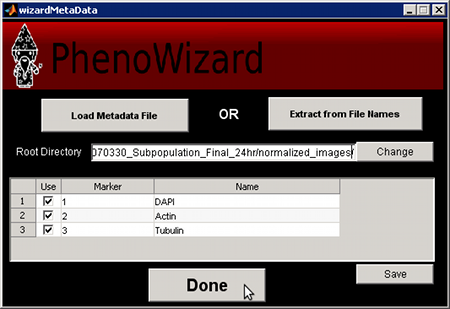
The Metadata Window Wizard will show up. Check that all the information is correct and
click on Done.
Using the File Structure
This option should be used only if no metadata file exists for your dataset. In this case,
PhenoRipper allows you to detect the organization of your images from the file structure.
Click on File Structure. A new window will popup.

Select the root directory that contains your data and select a file structure template matching your data. You will also have to change the File Extension and the Channel Separator if the default one doesn’t match your data filenames.
For our example, select xxx/Slack_PNAS_Data_Set_PNG/ as the Root Directory, the first template A1/xxxxx-1.png, png as the File Extension and - as the Channel Separator.
Then press Detect Channels to detect the different channels in your dataset and set their
names.
 In our example, Marker 1 is DAPI, Marker 2 is Tubulin and Marker 3 is Actin. Enter
these names into the table and press Next. You can also export the detected file structure
into a metadata file by clicking on Export Metadata File.
In our example, Marker 1 is DAPI, Marker 2 is Tubulin and Marker 3 is Actin. Enter
these names into the table and press Next. You can also export the detected file structure
into a metadata file by clicking on Export Metadata File.
If your data does not match a pre-defined PhenoRipper template, it is possible to add new
templates to match your file structure. Select the template that best matches your data and
click on Edit. A new window will pop up:
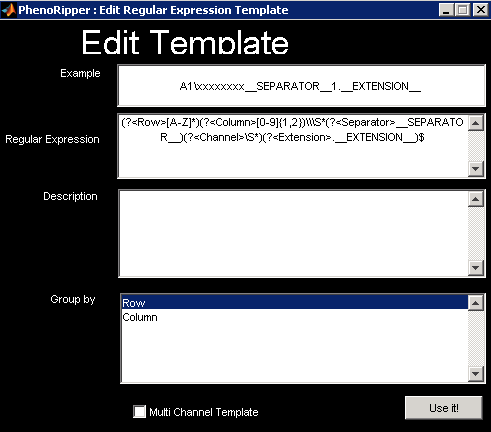 As you can see, templates are based on regular expressions. You need to provide a
regular expression and an example that matches the regular expression. The example will
be used as the name for your new template on the template list. Click on Use it! to use it
right away. This will close the Edit Template window and take you back to the
PhenoWizard. Here you should click on Save if you want to add this new template to
your default template list.
As you can see, templates are based on regular expressions. You need to provide a
regular expression and an example that matches the regular expression. The example will
be used as the name for your new template on the template list. Click on Use it! to use it
right away. This will close the Edit Template window and take you back to the
PhenoWizard. Here you should click on Save if you want to add this new template to
your default template list.
If you are familiar with regular expressions, you will notice that we use two unusual expressions, __SEPARATOR__ and __EXTENSION__. These expressions refer to the Separator and File Extension specified in the PhenoWizard and allow reuse of a template even if these parameters have been modified.
To test your regular expressions, you can use regexphelper, a tool available in the MATLAB file exchange at the following URL http://www.mathworks.com/matlabcentral/fileexchange/15215-regexphelper.
Learn Image Phenotypes
You now have to choose a strategy for sampling a representative subset of the data. By default, PhenoRipper chooses a random selection of images. However, users who wish to avoid sampling redundantly from the same condition have the option to let PhenoRipper know that certain images in a group are expected to be similar (like images taken from the same well or condition).
In our example, we have the option to not group ("Random"), group by Drug, or group by Drug class. Here, we will choose to group by Drug, which tells PhenoRipper that all replicate images for a single drug perturbation may be considered "equivalent" and that it should take representative images from across drugs (see FAQ Is PhenoRipper Supervised? for more details).

You should now see the Load Data Successfully dialog. Note that you have to wait for the computer to finish analyzing your file structure -the more images you have, the longer you have to wait.
You will then be taken back to the main interface where you can move to the next step.
Set Parameters
Click on Set Parameters to set the PhenoRipper parameters.
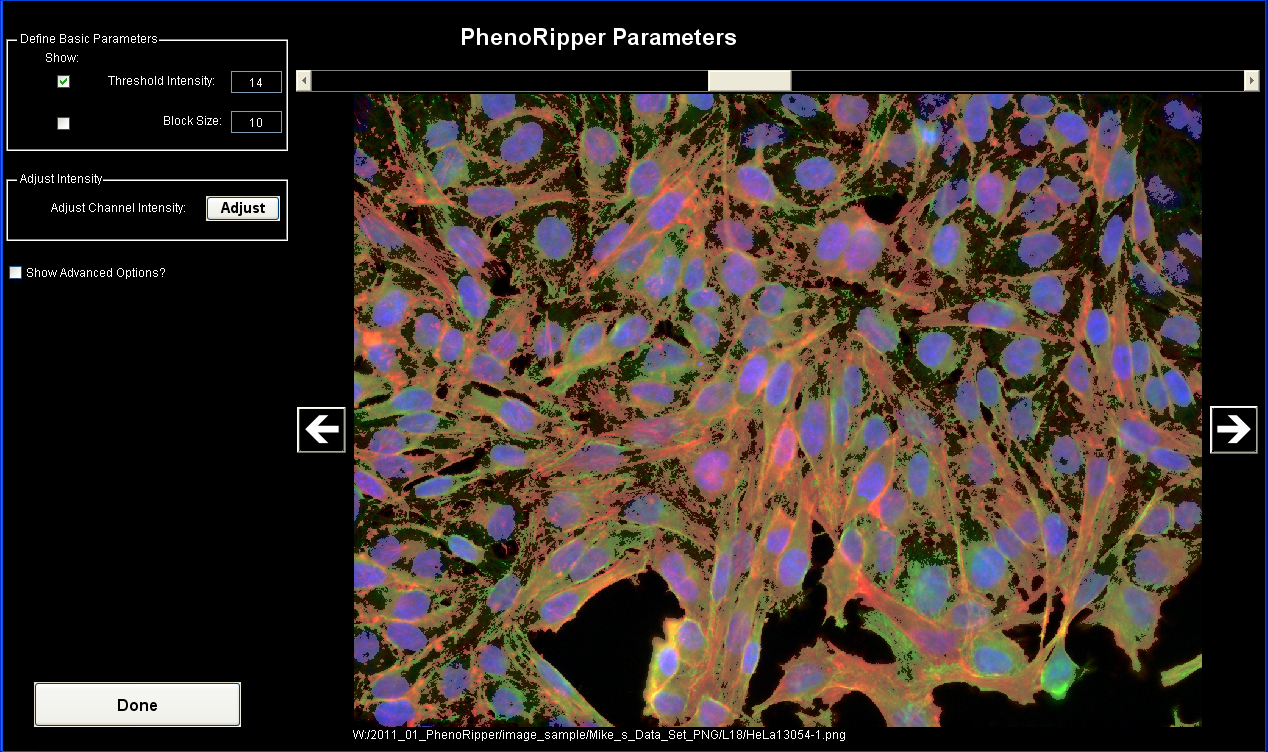
Visualize Parameters
Threshold value is automatically pre-calculated, but can be modified. By selecting Threshold Intensity in the Define Basic Parameters panel, you can visualize which part of the image will be analyzed (in brighter transparent gray). The rest (darker) will be considered background and will not be analyzed. In the current implementation of PhenoRipper, only one threshold intensity value will be set for the entire image dataset, so you should click on the left/right arrows to visualize other images and check if this threshold value is appropriate for them.
You can also tick the Block Size check box in the Define Basic Parameters panel to visualize the blocks. An average of 20-30 blocks per cell is recommended based on our own experience; after you gain experience with the program on your own datasets, you may wish to vary this parameter. The default value is 15 pixels, so you have to change it
PhenoRipper also offers you the ability to rescale channel by clicking on Adjust in the Adjust Intensity panel.
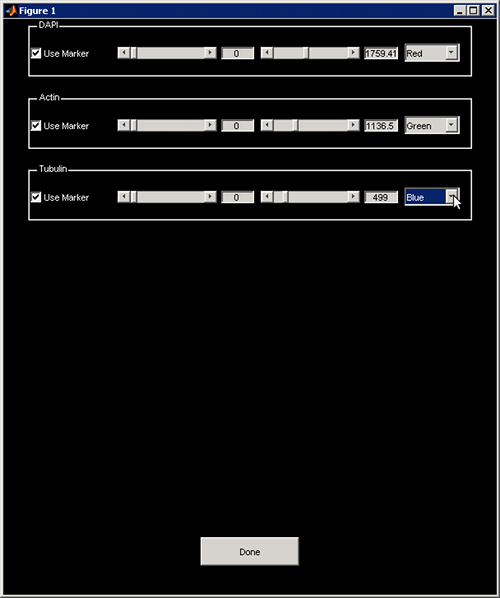
A new windows popup allows you to modify your channel color and intensity.
Advanced Parameters
Extra parameters are available by clicking on Show Advanced Options, but default values should work on the majority of the dataset.

Click on the button Done to go back to the main interface.
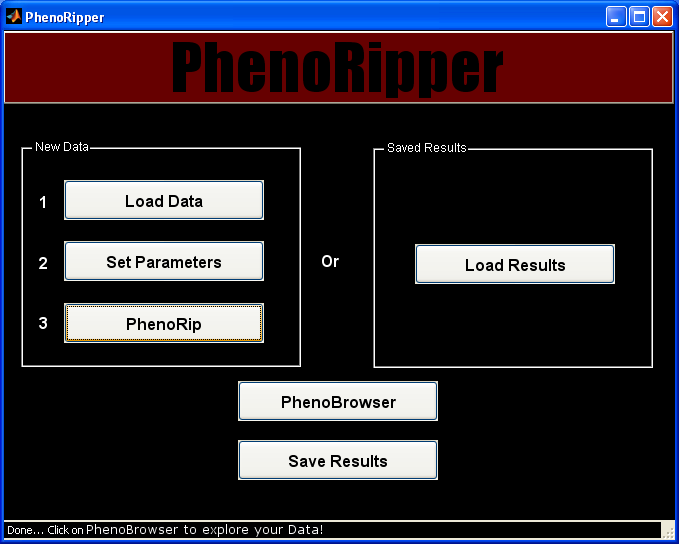
Run PhenoRipper
PhenoRip
Start the analysis by clicking on PhenoRip and wait for the end of the analysis.

Analysis will go through 3 stages:
- Learning Block Types
- Learning Superblock Types
- Analysis Images
Explore your Data
Overview
 You can click on some MDS points to view the corresponding image. Select Point 1 or Point 2 and an automatic comparison will be displayed showing some differences based on the block type.
You can click on some MDS points to view the corresponding image. Select Point 1 or Point 2 and an automatic comparison will be displayed showing some differences based on the block type.
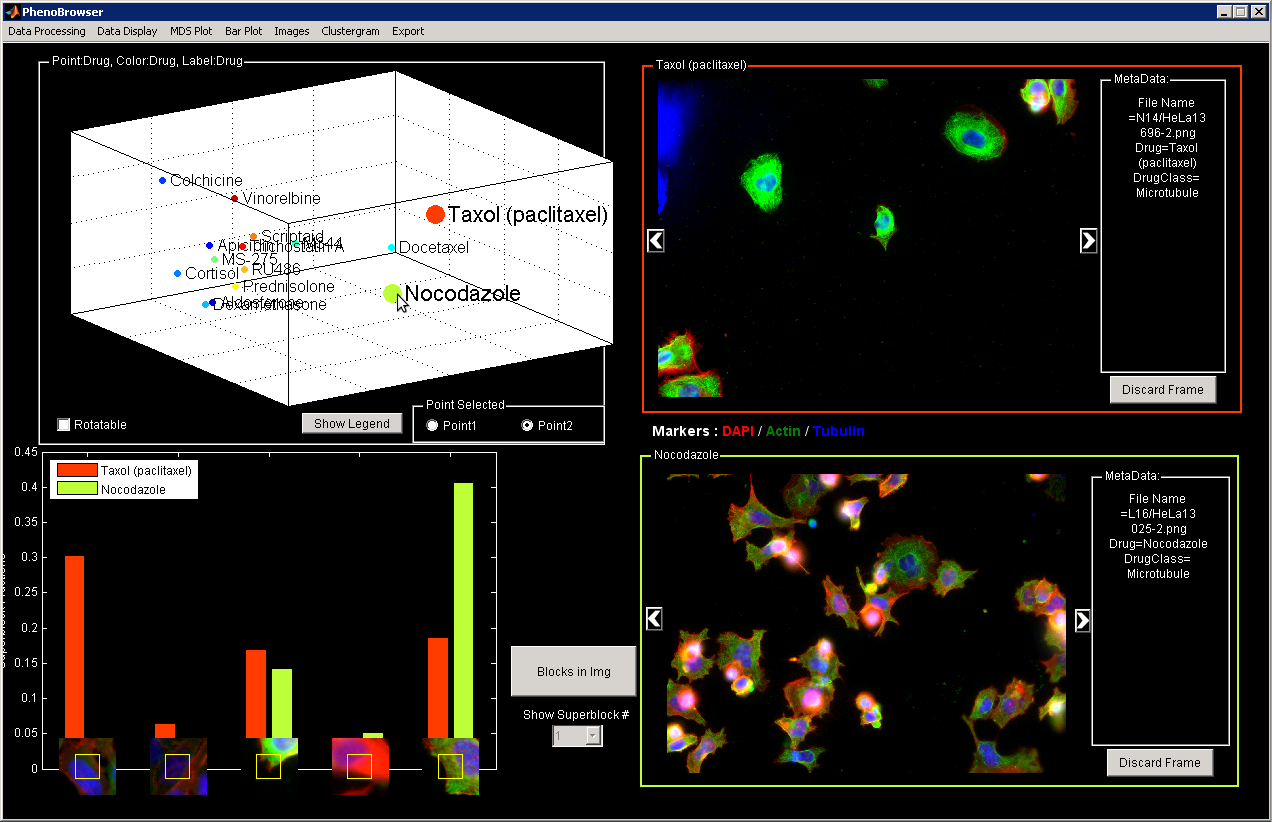 To rotate the MDS plot, click on Rotalable and drag and drop on the plot. Once you are satisfied of the position, you will need to un-select the Rotalable check box to be able to click on points and visualize the corresponding image.
To rotate the MDS plot, click on Rotalable and drag and drop on the plot. Once you are satisfied of the position, you will need to un-select the Rotalable check box to be able to click on points and visualize the corresponding image.

Adjust visualization
Discard Frame
You can remove automatically the empty frames.

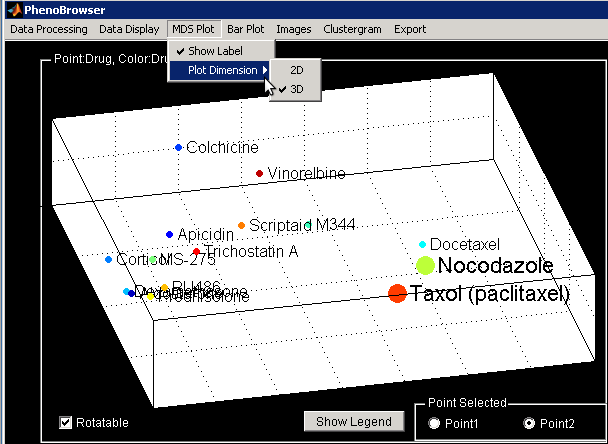
Change MDS Visualization
It is also possible to change the points color by group. Similarly, the labeling can also be
modified and even masked.

You can also show the best 2D separation by switching the Plot Dimension to 2D.
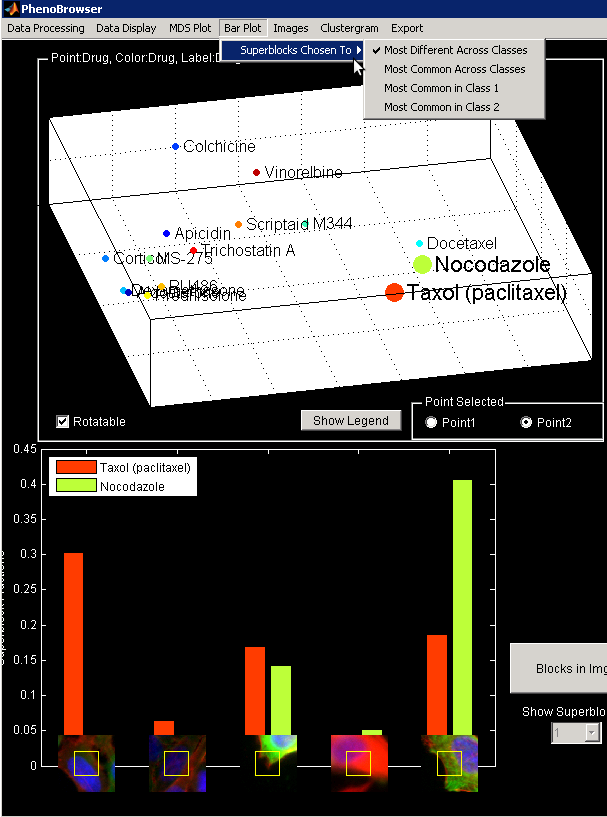
Change Bar Plot Visualization
The Bar plot can be adjusted to display different Superblocks according to different criteria.
Plot Clustergram
The Clustergram is accessible through the Explorer menu (Clustergran>Launch Clustergram).

This will launch a new window showing the Clustergram. Different option to visualize the clustergram are available through the menu.
The grouping of the data will be the same as the one used to plot the MDS.
